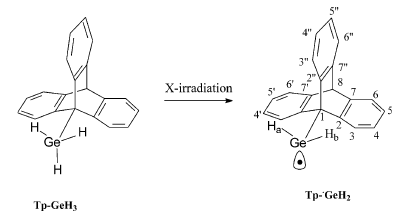
-
ESR/DFT study of bis-iminophosphorane cation radicals
A. Matni, L. Boubekeur, P. Grosshans, N. Mézailles, G. Bernardinelli, P. Le Floch and M. Geoffroy
Magnetic Resonance in Chemistry, 45 (12) (2007), p1011-1017


DOI:10.1002/mrc.2079 | unige:3578 | Abstract | Article PDF

Bis-iminophosphoranes containing various types of linkers between two R3P=N moieties were electrochemically oxidized at controlled potential in situ in the electron spin resonance (ESR) cavity. For linkers constituted of phenylenes, conjugated phenylenes or merely a dicyanoethylenic bond, this oxidation led to well-resolved ESR spectra which were characterized by their g values and by their 1H, 14N and 31P isotropic hyperfine constants. These coupling constants agree with those calculated by DFT for the corresponding cation radicals. Experimental and theoretical results clearly indicate that in these species the unpaired electron is mostly delocalized on the bridge and on the nitrogen atoms while the spin density on the phosphorus atoms is particularly small. Cyclic voltammetry and ESR spectra show that the nature of the bridge between the two iminophosphoranes considerably influences the oxidation potential of the compound as well as the stability of the radical cation. Information about the conformation of the precursor containing two Ph3P=N moieties separated by a âC(CN)=C(CN)âgroup was obtained from its crystal structure.